Hemocromatose hereditária (HH) é uma doença genética do metabolismo do ferro caracterizada por aumento da absorção intestinal e acúmulo progressivo do metal em diferentes órgãos. A mutação mais comum é no gene HFE. Nos Estados Unidos, cerca de 1 em 300 brancos não hispânicos tem hemocromatose hereditária, com taxas mais baixas entre outras raças e etnias.
No Brasil não há dados bem definidos. Embora o fígado seja o órgão mais comumente afetado clinicamente, a doença também tem manifestações endócrinas, em articulações e cardíacas.
Metabolismo do ferro: Um perfeito sincronismo entre absorção, utilização e estoque de ferro é essencial para a manutenção do equilíbrio desse metal no organismo. O ferro da dieta é absorvido pelos enterócito no intestino delgado proximal. Os principais tipos de ferro na dieta são ferro heme, que é prontamente absorvido, e ferro não heme, que é predominantemente ferro férrico.
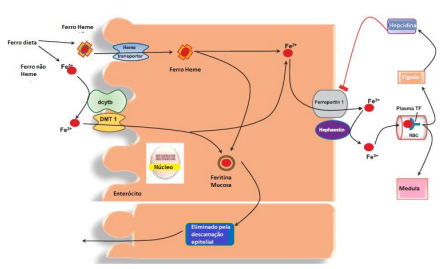
Para facilitar o transporte de ferro férrico insolúvel através da superfície luminal do enterócito, o ferro férrico (Fe3+) é reduzido pela redutase férrica duodenal citocromo B (dcytb) para ferro ferroso (Fe2+), que é então transportado para o enterócito por transportador de metal divalente (DMT1).
Uma vez dentro da célula, o ferro pode ser armazenado ligado à ferritina ou pode ser transferido através da superfície basolateral do enterócito por meio da proteína de transporte FPN (Ferroportina). O processo de exportação também envolve uma ferroxidase, que converte o ferro ferroso de volta em ferro férrico. Esta etapa é necessária para que o ferro se ligue a transferrina. A transferrina é a forma em que o ferro é entregue aos locais de sua utilização, como a medula óssea. A transferência de ferro através do enterócito é regulada pela hepcidina. A ligação da hepcidina ao FPN faz com que a última proteína seja internalizada e degradada no enterócito. A eliminação do transportador impede a saída de ferro da célula. O ferro retido no enterócito é então liberado quando a célula epitelial conclui o ciclo vital.

Manifestações Clínicas: A suspeita clínica de HH depende principalmente do conhecimento da doença, uma vez que não há uma manisfetação única ou típica. Artropatia, pigmentação da pele, diabetes mellitus, disfunção endócrina, cardiomiopatia, hipogonadismo, disfunção hepática, amenorreia, perda de libido, hipopituitarismo são manifestações da HH.

Porém, no início da doença são mais comuns a fadiga e artralgias. Estas manifestações ocorrem mais cedo nos homens do que nas mulheres, mais comumente na quarta ou quinta décadas de vida. Na menopausa já cessaram as perdas comuns do ferro devido a menstruação, gravidez e lactação, compensando o aumento da absorção de ferro durante esse período.
Genética: A HH está vinculada a variantes em diversos genes relacionados à regulação do ferro. A gravidade da variante causal correlaciona-se com o curso do tempo de sobrecarga de ferro e danos a órgãos. As variantes mais graves levam ao início dos sintomas em pacientes pediátricos, enquanto as variantes menos graves podem levar ao início dos sintomas em adultos ou podem nunca resultar em sintomas.
A forma mais comum de HH adulto tem sido associada a variantes (C282Y, H63D e S65C) do gene HFE, que codifica uma proteína responsável pela regulação do ferro. A HH pediátrica é rara. A maioria dos casos pediátricos está ligada a variantes do gene HFE2, que codifica a hepcidina.
A mutação H63D é mais comum do que C282Y e é encontrada na maioria das populações em todo o mundo, com a maior prevalência entre os brancos, dos quais aproximadamente 20% carregam -pelo menos 1 cópia de H63D. A mutação S65C é menos comum do que C282Y ou H63D, com uma frequência de heterozigoto de cerca de 2% entre brancos. Esta mutação parece ter um modesto efeito sobre o metabolismo do ferro na presença da mutação C282Y, mas doença relacionada à sobrecarga de ferro não foi relatada em C282Y / S65C heterozigotos compostos.
Nem o homozigoto, nem o heterozigoto para a mutação H63D ou S65C evoluem com sobrecarga de ferro patológica.
Segundo o Guideline do Am. J Gastroenterol 2019 duas recomendações são importantes:
“Recomendamos que os membros da família, principalmente parentes de primeiro grau de pacientes com diagnóstico de HH, devem ser rastreados (forte recomendação, qualidade moderada de evidência) ”
“Recomendamos que os indivíduos com a mutação H63D ou S65C na ausência de C282Y sejam informados de que eles não possuem risco aumentado de sobrecarga de ferro” (recomendação condicional, evidência de qualidade muito baixa). O motivo é que as sobrecargas de ferro por outras condições são mais comuns do que os outros tipos de hemocromatose não HFE.
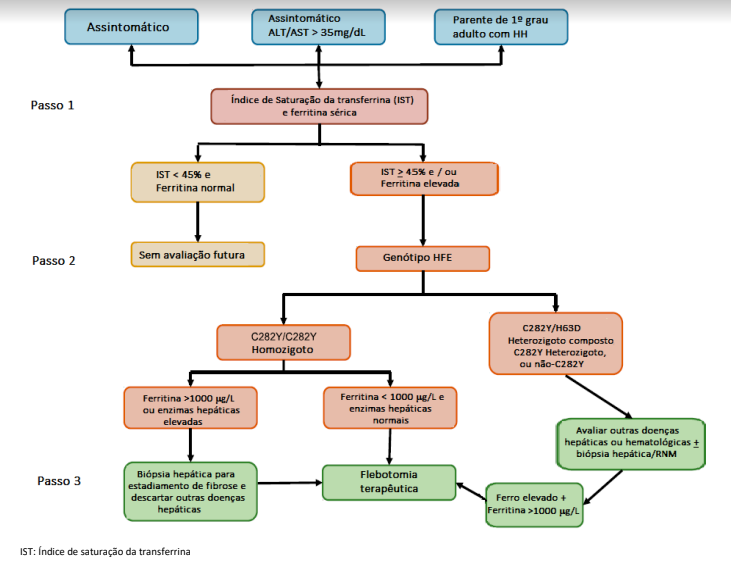
Avaliação Laboratorial: As abordagens iniciais para a avaliação de pacientes com suspeita de distúrbios da sobrecarga do ferro incluem dosagem do ferro sérico, índice de saturação da transferrina, ferritina e capacidade de ligação de ferro. O índice de saturação da transferrina é o teste de triagem inicial e o jejum não é necessário.

A sobrecarga de ferro também pode estar presente com um nível elevado de Ferritina e um nível normal de saturação da transferrina, particularmente em sobrecarga de ferro não relacionada a HFE.
A Ferritina é um excelente preditor de fibrose avançada, mas não tem especificidade como um teste de triagem, porque a hiperferritinemia pode estar presente em outras condições, incluindo doença hepática alcoólica, não alcoólica, HCV e doença neoplásica.
Em homozigotos C282Y, uma Ferritina acima de 1.000 ng/mL, associada a aminotransferase elevadas e plaquetopenia podem predizer a cirrose em mais de 80% dos pacientes.
Uma ferritina inferior a 200 ng/mL em mulheres na pré-menopausa ou 300 ng/mL em homens e mulheres pós-menopausa, em combinação com uma saturação da transferrina de 45%, tem um valor preditivo negativo de 97% para excluir sobrecarga de ferro. Vide algoritmo.
ALGORÍTMO HEMOCROMATOSE
Baseado nas evidências disponíveis , reproduzimos um algoritmo para o diagnóstico da hemocromatose, adaptado de The American College of Gastroenterology, ACG Clinical Guideline: Hereditary Hemochromatosis, disponível em: https://journals.lww.com/ajg/fulltext/2019/08000/acg_clinical_guideline__hereditary_hemochromatosis.11.aspx
Edição 09. Setembro/2021.
Assessoria Médica – Lab Rede
Referências
1. Kowdley, Kris V. MD, FACG1; Brown, Kyle E. MD, MSc2,3,4; Ahn, Joseph MD, MS, MBA, FACG (GRADE Methodologist)5; Sundaram, Vinay MD, MSc6 ACG Clinical Guideline: Hereditary Hemochromatosis, The American Journal of Gastroenterology: August 2019 – Volume 114 – Issue 8 – p 1202-1218 doi: 10.14309/ajg.0000000000000315




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}